On June 16, 2025, TransMIT GmbH dedicated itself to EU regulation as part of the GO-Bio initial seminar series and invited participants to two online presentations.
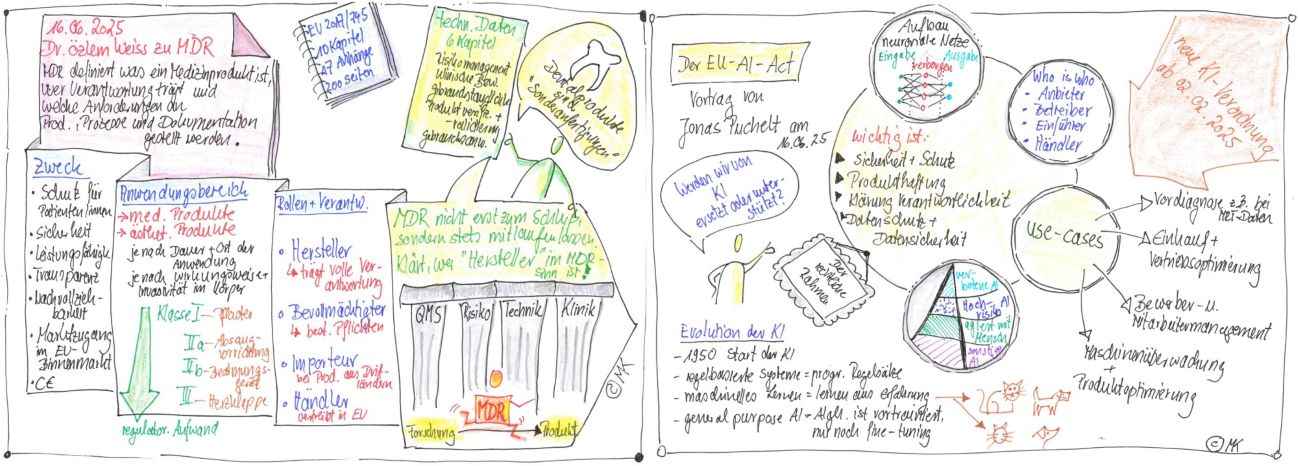
In the first presentation, Dr. Özlem Weiss introduced the MDR topic, presented the most important relevant regulations and defined what a medical device is, who is responsible and what requirements are placed on products, processes and documentation. Dr. Özlem Weiss is Managing Director of Expertants GmbH, a service provider for product development and regulatory services for medical devices and additive manufacturing. She coaches and evaluates start-ups in the life sciences sector.
Right at the beginning of her presentation, she raised awareness of the actual purpose of the complex regulatory system, above all for the protection and safety of patients, as well as for the guarantee of using a high-performance product. Transparency and traceability in the approval process must be ensured and market access to the EU internal market must be regulated. Dr. Weiss referred to the application of the MDR regulations for the approval of all medical products, but also aesthetic products without a medical effect. Many participants already suspected that the regulatory burden increases depending on the duration and place of use and depending on the mode of action and invasiveness in the body, but the exact classification of medical devices into Class I, IIa, IIb and III was not familiar to many, nor was the fact that dental products are often custom-made and follow a special approval path.
The speaker presented the entire regulatory and CE marking process, highlighted the complex and underestimated stages and motivated the participants not to place these important regulatory requirements at the end of product development, but to keep the regulatory process running throughout the entire development period. It is particularly important to be clear right from the start who the manufacturer of a product is in terms of the MDR, because the manufacturer bears full responsibility for the product.
It was particularly important to Dr. Weiss to present the four pillars: quality management, risk management, technology and clinic, and to discuss the associated documentation. For her, however, documentation is not the goal of product development but the link between development, safety and certification. At the end of this very interesting presentation, Dr. Weiss addressed various questions from the participants and reported on her own experiences in supporting start-up teams and their product approvals.
In the second presentation, Mr. Jonas Puchelt, lawyer from the commercial law firm FPS in Frankfurt, presented the legal framework of the EU AI Act and pointed out similarities in EU legislation as a transition from the first presentation. As a specialist lawyer for information technology law and data protection officer (DSC), Mr. Puchelt is familiar with inquiries regarding the use of artificial intelligence (AI) in product development. He started with the question of whether we are being replaced or supported by AI.
In addition to clarifying what neural networks, machine learning and artificial intelligence are and what distinguishes them, he focused on how the new AI regulation of 02/02/2025 relates to safety and protection, what product liability means, who bears responsibility and what needs to be considered in terms of data protection and data security.
Highly illustrative use cases made the various areas of application of AI clear to the participants. For example, AI is already being used in machine monitoring and product optimization, in applicant and employee management, in purchasing and in sales optimization. It became particularly interesting for the participants when Mr. Puchelt discussed the use of AI in pre-diagnosis, e.g. from MRI data or other personal medical data. This area is currently the subject of much discussion in the medical community and product developments are aimed at it.
Mr. Puchelt went into detail about the classification of high-risk AI systems and what obligations users and operators have.
It was also very important to him to sensitize the participants to keep these AI regulations in mind at an early stage of product development and to clarify whether, for example, as a manufacturer of a medical app, you are a provider, operator, importer or distributor. A provider is anyone who develops an AI system or has one developed. An operator is someone who uses an AI system under their own responsibility. An importer is anyone who places an AI system from a third country on the market in the EU. Distributor is anyone who is part of a supply chain and makes an AI system available on the EU market.
He emphasized that AI in itself is not responsible for misconduct and that data protection and data security must also be ensured.
Before moving on to the discussion, Mr. Puchelt addressed the consequences of non-compliance with the new AI Directive, which includes fines, claims for damages by injured parties and even class actions.
At the end of this very clear presentation, he invited the participants to carry out their own check before or during product development at fps-law.de/de/FPS-Ki-check „Was müssen Sie beim Einsatz von KI beachten?