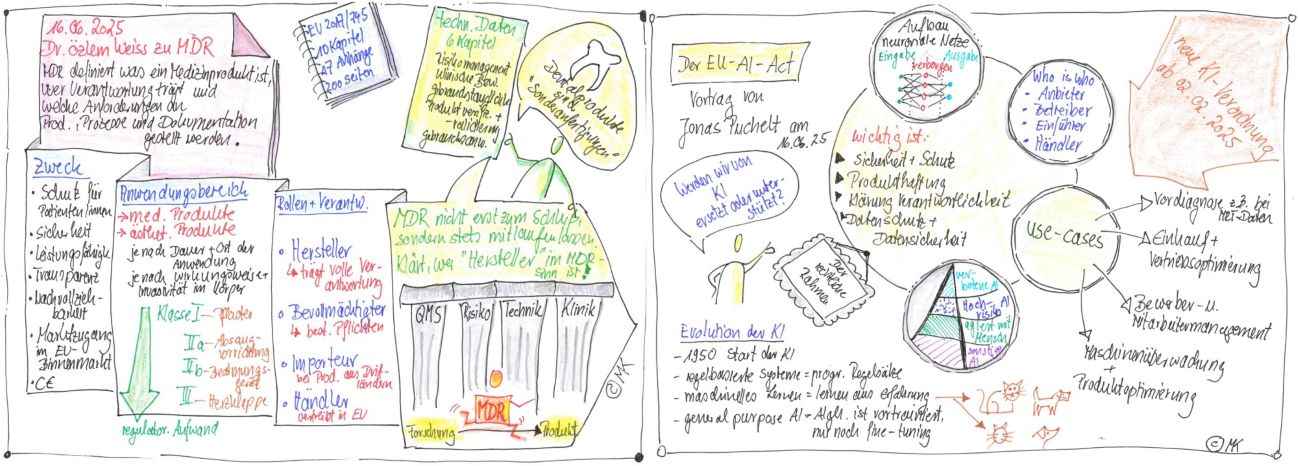
Am 16.06.2025 widmete sich die TransMIT GmbH im Rahmen der GO-Bio initial Seminarreihe der EU-Regulatorik und lud zu zwei online Vorträgen ein.
Im ersten Vortrag führte Frau Dr. Özlem Weiss in die MDR-Thematik ein, stellte die wichtigsten einschlägigen Regelwerke vor und definierte was ein Medizinprodukt ist, wer Verantwortung trägt und welche Anforderungen an Produkte, Prozesse und die Dokumentation gestellt werden. Dr. Özlem Weiss ist Geschäftsführerin der Expertants GmbH, einem Dienstleister für Produktentwicklung und regulatorische Dienstleistungen für Medizinprodukte und additive Fertigung. Sie coacht und evaluiert Start-ups aus den Bereichen Life Sciences.
Gleich zu Beginn ihres Vortrages sensibilisierte sie für den eigentlichen Zweck der aufwendigen Regulatorik, vor allem für den Schutz und die Sicherheit der Patientinnen und Patienten, sowie für die Garantie ein leistungsfähiges Produkt zu verwenden. Transparenz und Nachvollziehbarkeit bei der Zulassung müssen gegeben und der Marktzugang in den EU-Binnenmarkt geregelt sein. Frau Dr. Weiss verwies auf die Anwendung der MDR-Regulatorik für die Zulassung aller medizinischen Produkte, aber auch der ästhetischen Produkte ohne medizinische Wirkung. Dass der regulatorische Aufwand je nach Dauer und Ort der Anwendung und je nach Wirkungsweise und Invasivität im Körper steigt, vermuteten viele Teilnehmenden bereits, aber die genaue Einteilung der Medizinprodukte in Klasse I, IIa, IIb und III, war vielen so nicht geläufig, ebensowenig, wie die Tatsache, dass Dentalprodukte oft Sonderanfertigungen sind und einen besonderen Zulassungsweg einschlagen.
Die Referentin stellte den gesamten Weg der Regulatorik und CE-Kennzeichnung vor, ging besonders auf aufwendige und unterschätzte Etappen ein und motivierte die Teilnehmenden dazu, diese wichtige Regulatorik nicht an das Ende einer Produktentwicklung zu stellen, sondern die Regulatorik während der gesamten Entwicklungszeit mitlaufen zu lassen. Besonders wichtig sei es, sich gleich zu Beginn darüber im Klaren zu sein, wer der Hersteller eines Produktes im Sinne des MDR ist, weil der Hersteller die volle Verantwortung für das Produkt trägt.
Es war Frau Dr. Weiss besonders wichtig, die vier Säulen: Qualitätsmanagement, Risikomanagement, Technik und Klinik vorzustellen und auf die dazu gehörenden Dokumentationen einzugehen. Für sie ist die Dokumentation allerdings nicht das Ziel einer Produktentwicklung sondern das Bindeglied zwischen Entwicklung, Sicherheit und Zertifizierung. Am Ende dieses sehr interessanten Vortrags ging Frau Dr. Weiss noch auf verschiedene Fragestellungen der Teilnehmenden ein und berichtete von eigenen Erfahrungen bei der Begleitung von Gründerteams und deren Produktzulassungen.
Im zweiten Vortrag stellte Herr Jonas Puchelt, Rechtsanwalt von der Wirtschaftskanzlei FPS aus Frankfurt den rechtlichen Rahmen des EU AI-Act vor und wies zur Überleitung aus dem ersten Vortrag gleich auf Übereinstimmungen bei der EU-Gesetzgebung hin. Als Fachanwalt für Informationstechnologierecht und Datenschutzbeauftragter (DSC) ist Herr Puchelt vertraut mit Anfragen zur Nutzung der Künstlichen Intelligenz KI, Artificial Intelligence AI bei der Produktentwicklung. Er startete gleich mit der Frage, ob wir von der KI ersetzt oder unterstützt werden.
Neben der Begriffsklärung, was neuronale Netze, maschinelles Lernen und Künstliche Intelligenz sind und was sie auszeichnet, ging er vor allem darauf ein, wie es sich jetzt mit der neuen KI-Verordnung vom 02.02.2025 mit Sicherheit und Schutz verhält, was Produkthaftung bedeutet, wer die Verantwortung trägt und was beim Datenschutz und der Datensicherheit zu beachten ist.
Sehr anschauliche use-cases machten den Teilnehmenden die verschiedenen Einsatzgebiete der KI deutlich. So wird KI bereits bei der Maschinenüberwachung und der Produktoptimierung eingesetzt, im Bewerber- und Mitarbeitermanagement, im Einkauf und bei der Vertriebsoptimierung. Besonders interessant wurde es für die Teilnehmenden, als Herr Puchelt auf den Einsatz der KI bei Vordiagnosen z. B. aus MRT-Daten oder anderen persönlichen medizinischen Daten einging. Dieser Bereich wird derzeit sehr stark in der medizinischen Community diskutiert und Produktentwicklungen zielen darauf ab.
Herr Puchelt ging ausführlich auf die Klassifizierung von Hochrisiko-KI-Systemen ein und welche Pflichten auf Nutzer und Betreiber zukommen.
Auch ihm war es sehr wichtig, die Teilnehmenden zu sensibilisieren, diese KI-Regulatorik frühzeitig bei der Produktentwicklung im Blick zu haben und zu klären, ob man z.B. als Hersteller einer medizinischen App, Anbieter, Betreiber, Einführer oder Händler ist. Anbieter ist, wer ein KI-System entwickelt oder entwickeln lässt. Betreiber ist, wer ein KI-System in eigener Verantwortung verwendet. Einführer ist, wer ein KI-System aus einem Drittland in der EU in Verkehr bringt. Händler ist, wer Teil einer Lieferkette ist und ein KI-System auf dem EU-Markt bereitstellt.
Er betonte, dass die KI an sich nicht für Fehlverhalten verantwortlich ist und auch der Datenschutz und die Datensicherheit sichergestellt werden müssen.
Bevor es in die Diskussion überging, ging Herr Puchelt noch auf die Folgen, der Nichtbeachtung der neuen KI-Richtlinie ein, die Geldbußen, Schadensansprüche Geschädigter und sogar Verbandsklagen umfasst.
Zum Abschluss dieses sehr anschaulichen Vortrages lud er die Teilnehmenden noch ein, vor oder während der Produktentwicklung selber einen Check unter fps-law.de/de/FPS-Ki-check zu machen „Was müssen Sie beim Einsatz von KI beachten?